Research teams led by Prof. YANG Huangtian from the Shanghai Institute of Nutrition and Health (SINH), Chinese Academy of Sciences (CAS) and Prof. YIN Huiyong from City University of Hong Kong, have unveiled the critical role of mitochondrial uncoupling protein 3 (UCP3) against pathological cardiac hypertrophy via downregulation of aspartate metabolism. Entitled “Uncoupling protein 3 protects against pathological cardiac hypertrophy via downregulation of aspartate”, the study was published online on Journal of Molecular and Cellular Cardiology, an official journal of the International Society for Heart Research, on March 2, 2025.
Pathological cardiac hypertrophy, a hallmark of pressure overload-induced heart diseases such as hypertension and aortic stenosis, is closely linked to mitochondrial dysfunction, metabolic remodeling, and dysregulation of tricarboxylic acid (TCA) cycle intermediates. UCP3, an inner mitochondrial membrane anion transporter, has been shown by Prof. YANG Huangtian’s group to protect cardiomyocytes from ischemic injury by binding to mitochondrial permeability transition pore regulatory proteins, while its role in pressure overload-induced cardiac hypertrophy remains unclear.
The research team observed significant downregulation of UCP3 in both a mouse model of transverse aortic constriction (TAC)-induced cardiac hypertrophy and norepinephrine-treated neonatal rat cardiomyocytes (NRCMs). Using systemic and cardiomyocyte-specific UCP3 knockout (UCP3cKO) mice, they demonstrated that UCP3 deficiency exacerbated TAC-induced cardiac hypertrophy and cardiac dysfunction. Conversely, cardiomyocyte-specific UCP3 overexpression (UCP3cOE) alleviated these pathological phenotypes. In vitro studies further confirmed that UCP3 knockdown aggravated norepinephrine-induced cardiomyocyte hypertrophy, while UCP3 overexpression suppressed it.
Mechanistically, UCP3 interacted with glutamic-oxaloacetic transaminase 2 (GOT2) to regulate aspartate metabolism. In TAC-induced hypertrophic hearts, GOT2 activity and aspartate levels were markedly elevated, which were further exacerbated in UCP3cKO mice and reversed in UCP3cOE mice. Similarly, in norepinephrine-treated NRCMs, UCP3 knockdown increased the elevation of GOT2 activity, while overexpression of UCP3 attenuated the elevation, accompanied with a decrease in the aspartate level. Furthermore, norepinephrine stimulation disrupted the endogenous binding between UCP3 and GOT2 in NRCMs, while exogenous aspartate supplementation abolished the protective effects of UCP3 overexpression against cardiomyocyte hypertrophy. These results indicate that UCP3 suppresses pathological cardiac hypertrophy by inhibiting GOT2-mediated aspartate accumulation.
In summary, this study elucidated the protective role of UCP3 in pressure overload-induced cardiac hypertrophy and highlighted the central role of aspartate metabolism in UCP3-mediated cardioprotection. The findings also provided a theoretical foundation for developing metabolism-targeted therapeutic strategies to combat pathological cardiac hypertrophy.
The research was supported by grants from the National Natural Science Foundation of China and the Research Grants Council of Hong Kong, with additional support from the Analytical Testing and Experimental Animal Technology Platforms at SINH.
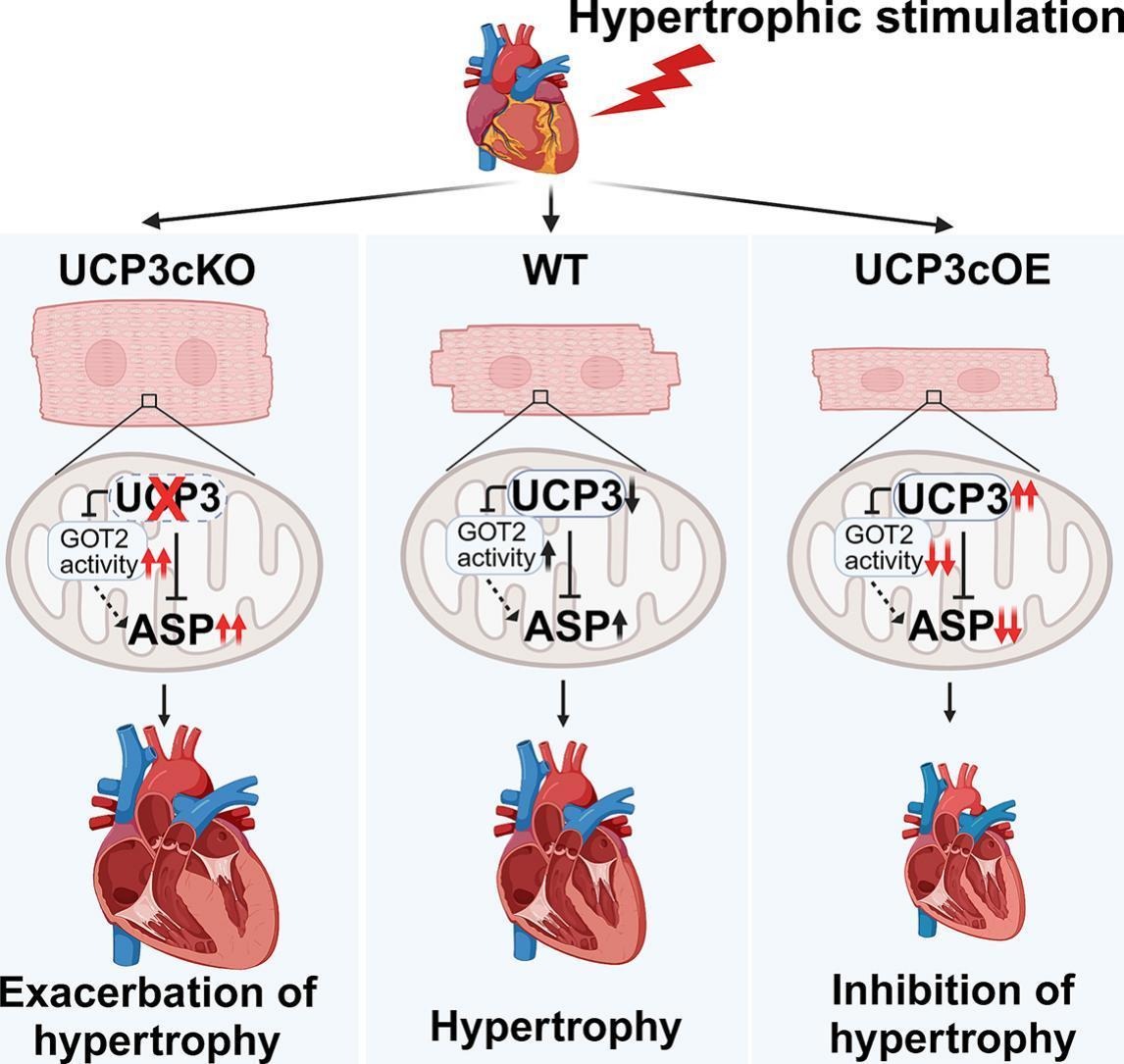
The proposed model for the protective effect of UCP3 against pathological cardiac hypertrophy via inhibiting aspartate accumulation. (Image provided by Prof. YANG Huangtian's team)
Media Contact:
WANG Jin
Shanghai Institute of Nutrition and Health,
Chinese Academy of Sciences
Email: wangjin01@sinh.ac.cn
Web: http://english.sinh.cas.cn/
