A recent study published in Neuroscience Bulletin demonstrated that missense mutations in PiT2-loop7 causes inorganic phosphate (Pi) dyshomeostasis by affecting the phosphorylation-regulated cell-surface localization of PiT2. This study was jointly completed by Dr. LIU Jing yu’s lab and ZHU Shujia’s lab at the Institute of Neuroscience (ION), CAS Center for Excellence in Brain Science and Intelligence Technology (CEBSIT) , Chinese Academy of Sciences. This work revealed the importance of the phosphorylation of PiT2-loop7 in the physiological functions of PiT2 and uncovered the underlying mechanisms by which the missense mutations in PiT2-loop7 lead to primary familial brain calcification (PFBC), opening up a new avenue of a research in PFBC therapy.
PFBC is an inheritable neurodegenerative disease mainly characterized by symmetrical calcification in the basal ganglia and/or other brain regions. The most common clinical manifestations of the patients include movement disorders, cognitive impairments, psychiatric signs, and headaches. Until now, there has been no effective treatment for PFBC. PiT2, encoded by SLC20A2, is capable of transporting extracellular Pi into the cell to maintain Pi homeostasis in the presence of Na+. Loss-of-function mutations of SLC20A2 were linked to PFBC, and it was proposed that PFBC is caused by Pi dyshomeostasis.
PiT2 mainly consists of two ProDom (PD) domains and a large intracellular loop region (PiT2-loop7). The PD domains are crucial for the Pi transport, but the role of PiT2-loop7 remains unclear. To date, about 147 mutations of SLC20A2 have been identified in PFBC patients. Although most of these mutations are located in the PD domains, some mutations are identified in PiT2-loop7, the majority of which are nonsense or frameshift mutations that probably cause PFBC due to C-PD1131 deletion. It has been suggested that the PiT2-loop7 domain might not participate in Pi transport, so how did these missense mutations in this domain (p.S283R, p.R382Q, p.T390A, p.H399P, and p.S434W) lead to PFBC?
To explore the detailed molecular mechanisms by which these missense mutations in PiT2-loop7 cause PFBC, the LIU’s team selected p.T390A and p.S434W in the PiT2-loop7 and p.S121C and p.S601W in the PD domains as representatives for investigation. Radioactive Pi uptake and electrophysiological data showed that the missense mutations in PiT2-loop7, similar to those in PD domains, could decrease the Pi transport activity of PiT2. Affinity test showed that the p.T390A and p.S434W mutations in PiT2-loop7 did not affect the substrate-binding ability of PiT2, while the p.S121C and p.S601W mutations in the PD domains affected the Pi but not the Na+-binding of PiT2. Combined with bioinformatics analysis, immunoblotting and immunofluorescence, the authors concluded that the p.T390A and p.S434W mutations in PiT2-loop7 affected the phosphorylation of PiT2 mediated by AMPK and AKT, respectively, thereby obstructing PiT2 transport to the cell surface. In PFBC patients, the other missense mutations in PiT2-loop7 (p.S283R, p.R382Q, and p.H399P) could also interfered with PiT2 phosphorylation and membrane localization, reducing the Pi transport activity of PiT2, and leading to PFBC.
In this study, the authors demonstrated that the missense mutations in PiT2-loop7 that cause PFBC could affect the phosphorylation of PiT2-loop7 and result in decreased cell surface expression of PiT2, eventually leading to a reduction in Pi transport; the p.S121C and p.S601W mutations in the PD domains could impair the Pi-binding ability of PiT2, thereby resulting in PiT2 dysfunction (Figure 1). This work improves our understanding of PFBC pathogenesis and provides theoretical bases for the development of PFBC therapies.
SUN Hao (College of Life Science and Technology, Huazhong University of Science and Technology) and postdoctoral scholar XU Xuan (CEBSIT), who were supervised by Dr. LIU Jingyu and ZHU Shujia, contributed equally to this work. This work received great help from Dr. XIONG Bo (Tongji Medical College, Huazhong University of Science and Technology) and was supported by the National Natural Science Foundation of China, Ministry of Science and Technology of the People’s Republic of China, and Shanghai Municipal Government.
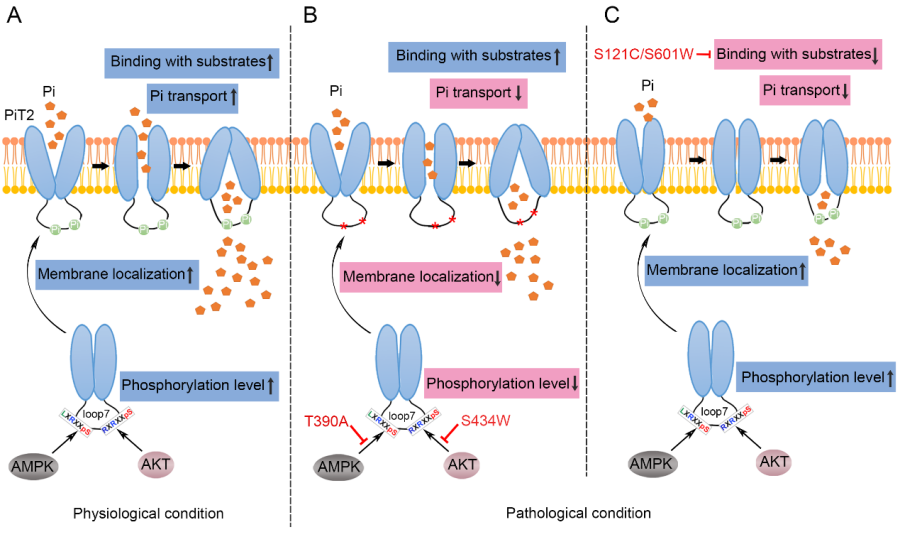
Figure 1. Proposed model of how different mutations in PiT2 cause Pi dyshomeostasis. (A) Under physiological conditions, during Pi transport, activated AMPK or AKT binds to PiT2 and induces phosphorylation, eventually promotes PiT2 transport to the cell surface and PiT2 function. (B, C) Under pathological conditions, the p.T390A and p.S434W mutations in PiT2-loop7 affect the phosphorylation-regulated membrane localization of PiT2, ultimately reduce Pi transport (B); the p.S121C and p.S601W mutations in the PD domains impair the Pi-binding of PiT2, thereby contributing to PiT2 dysfunction (C). The curved thin black arrows indicate the transport of PiT2 to the cell membrane, the thin black arrows starting from AMPK or AKT indicate the sites where they mediate PiT2 phosphorylation, and the red lines with bar ends indicate the inhibition of the physiological process.
https://doi.org/10.1007/s12264-022-00893-y
Keywords: PiT2-loop7, Mutation, Primary familial brain calcification, Phosphorylation, Pi dyshomeostasis
AUTHOR CONTACT:
LIU Jingyu
Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China.
E-mail: liujy@ion.ac.cn
